Hemofilie A en B & de stollingscascade
Hemofilie A en B: gelijkenissen en verschillen
In het algemeen, wanneer iemand zich verwondt, zal het bloed binnen enkele minuten stollen, waardoor de aanzet wordt gegeven voor de genezing. Het bloed van personen die lijden aan hemofilie stolt niet zoals het hoort omdat een van de stoffen die noodzakelijk is voor de stolling ontbreekt of in een te kleine hoeveelheid aanwezig is.
Personen die kampen met hemofilie A hebben een tekort aan het stollingseiwit factor VIII (factor acht). Deze factor kan volledig ontbreken of onvoldoende aanwezig zijn.Bij personen die lijden aan hemofilie B (of Christmas disease) is er een tekort aan het stollingseiwit factor IX (factor negen). Deze bloedaandoening lijkt sterk op hemofilie A, maar komt veel minder vaak voor.
Beide vormen van hemofilie kunnen enkel door een bloedonderzoek van elkaar onderscheiden worden. Inderdaad, hemofilie A en B veroorzaken dezelfde symptomen. Het onderscheid kan alleen worden gemaakt door de bloedplasmaconcentratie van de verschillende stollingsfactoren (VIII en IX) te onderzoeken.
Hemofilie A en B zijn zeldzaam: hemophilie A wordt vastgesteld bij 1 op de 10.000 jongens.Hemofilie B komt voor bij 1 op de 30.000 jongens. De groep hemofiliepatiënten bestaat voor 80% uit hemofilie A en voor 20% uit hemofilie B-patiënten.
De stollingscascade
Bij de bloedstolling spelen diverse stoffen een rol. Het merendeel ervan zijn eiwitten. Ons lichaam produceert deze stoffen voortdurend om steeds voorbereid te zijn en actie te kunnen ondernemen wanneer we ons verwonden. Naarmate de stoffen aangemaakt worden en vrijkomen in het bloed, vervangen ze de stoffen die eerder geproduceerd werden en nu ofwel gerecycleerd ofwel verwijderd worden. Meestal zijn er slechts kleine hoeveelheden van deze actieve stoffen aanwezig in ons bloed, maar wanneer we ons verwonden, wordt de productie snel verhoogd. Het bloed brengt de stoffen vervolgens naar de locatie van de wonde, waar ze de schade beperken en het genezingsproces activeren.
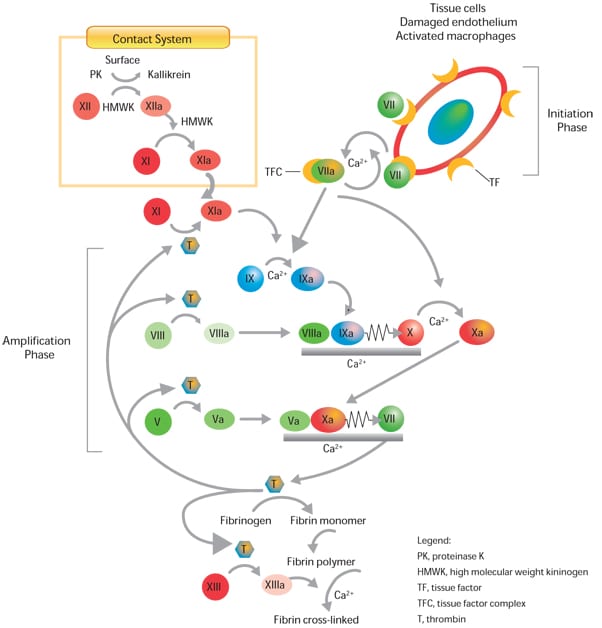
De meeste van deze stoffen worden factoren genoemd. Er zijn in totaal 12 hoofdfactoren, waaronder factor VIII.
Wanneer het lichaam onvoldoende factor VIII produceert, spreken we van hemofilie A.
Stollingsfactoren en aanverwante stoffen
(a: actieve vorm van de factor):
|
I |
fibrinogen (Ia: fibrine of klonter) |
|
II |
protrombine (IIa: trombine) |
|
V |
proaccelerine of labile factor |
|
VII |
proconvertine |
|
VIII |
antihemofilie factor - AHF |
|
IX |
PTC-Plasma Tromboplastinecomponent of Christmas Factor |
|
X |
Stuart-Prowerfactor of trombokinase |
|
IX |
PTA-Plasmatromboneoplastineantecedent |
|
X |
Stuart-Prowerfactor of trombokinase |
|
XI |
PTA-Plasmatromboneoplastineantecedent |
|
XII |
Hageman factor |
|
XIII |
firbine stabiliserende factor |
|
PL |
bloedplaatjes fosfolipide |
|
Ca** (IV) |
calcium Weefselfactor of weefseltromboplastine |
Begin jaren 60 ontdekten wetenschappers dat heel wat eiwitten een rol spelen bij de bloedstolling. Ze identificeerden en benoemden 12 verschillende stollingsfactoren en beschreven het stollingsproces in detail. In 1962 kende een internationaal comité een Romeins getal toe aan elk van deze eiwitten om verwarring te vermijden. De factoren die hemofilie A en B veroorzaken, werden respectievelijk factor VIII en factor IX genoemd.
In 1964 werd in het wetenschappelijke tijdschrift Nature een model gepubliceerd voor de wisselwerking tussen deze factoren, de zogenaamde bloedstollingscascade.
Hoewel het bloed van personen die lijden aan hemofilie niet stolt zoals het hoort, bloeden deze personen niet sneller of overvloediger dan anderen. De bloeding zal alleen langer duren. Zelfs in ernstige gevallen zal het bloed stollen, alleen verloopt het proces veel trager. De aandoening zorgt er meestal voor dat de klonter niet bestand is tegen de druk van het ontsnappende bloed. Dit zorgt ervoor dat de klonter niet hard kan worden, wat een negatieve impact heeft op het genezingsproces van de wonde.
