Hémophilie A et B & la cascade de coagulation
Hémophilie A et B
En général, lorsqu'une personne se blesse, un caillot de sang se forme en quelques minutes, ce qui amorce le processus de cicatrisation. Le sang d'une personne hémophile ne coagule pas normalement, car une des substances nécessaires à la formation d'un caillot est absente ou présente en quantité insuffisante. Chez les personnes hémophiles A, une protéine de coagulation appelée facteur VIII (facteur huit) est absente. Ce facteur peut être complètement absent ou présent en quantité insuffisante.
Chez les personnes atteintes d'hémophilie B (parfois appelée maladie de Christmas), une protéine de coagulation appelée facteur IX (facteur neuf) est absente ou présente en quantité insuffisante. Ce trouble hémorragique est très semblable à l'hémophilie A, mais est beaucoup moins courant.
Seul un examen sanguin peut distinguer les deux formes d'hémophilie. En effet, les hémophilies A et B présentent les mêmes symptômes. La distinction ne pourra se faire qu'en mesurant la concentration en facteur (VIII et IX) dans le plasma sanguin.
L'hémophilie A et B sont des maladies rares: l'hémophilie A se déclare chez 1 garçon sur 10.000. Pour l'hémophilie B, il s'agit d'un garçon sur 30.000. La communauté hémophile est composée pour 80% de patients hémophiles A et 20% présentant de l'hémophilie B.
La cascade de coagulation
Plusieurs substances jouent un rôle dans la coagulation sanguine. La plupart sont des protéines. L'organisme produit constamment ces substances, afin d'être toujours prêt à passer à l'action en cas de blessure. À mesure que ces substances sont produites et libérées dans le sang, elles remplacent celles qui avaient été produites avant, ces dernières étant soit recyclées, soit éliminées. Normalement, de faibles concentrations de ces substances actives sont présentes dans le sang, mais quand une blessure survient, il y a une augmentation rapide de la production de ces substances et le sang les transporte au site de la blessure, où elles limitent les dommages et amorcent la cicatrisation.
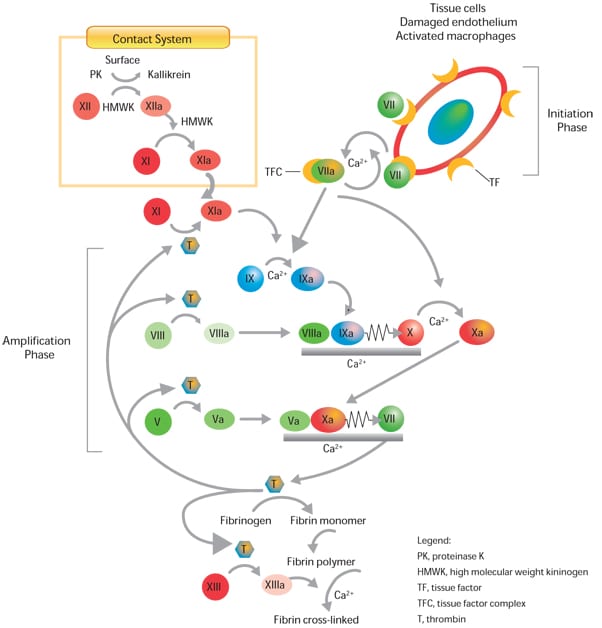
La plupart des substances sont appelées facteurs. Il y a en tout 12 facteurs principaux en tout, dont le facteur VIII.
Quand l'organisme ne produit pas la quantité normale de facteur VIII, on parle d'hémophilie A.
Facteurs intervenant dans la cascade de réactions de la coagulation
(a: forme active du facteur):
|
I |
fibrinogène (Ia : fibrine) |
|
II |
protrombine (IIa: trombine) |
|
V |
proaccélérine |
|
VII |
proconvertine |
|
VIII |
facteur antihémophilique A |
|
IX |
facteur antihémophilique B |
|
X |
facteur Stuart |
|
IX |
PTA = Plasma Thromboplastin Antecedent |
|
X |
Stuart-Prowerfactor of trombokinase |
|
XI |
PTA-Plasmatromboneoplastineantecedent |
|
XII |
facteur Hageman ou facteur contact |
|
XIII |
facteur stabilisant de la fibrine |
|
PL |
phospholipide plaquettaire |
|
Ca** (IV) |
calcium |
Au début des années 1960, les chercheurs ont découvert que de nombreuses protéines jouent un rôle dans la voie de la coagulation, ont identifié et nommé les 12 facteurs de coagulation, et ont décrit en détail le processus de coagulation.
En 1962, un comité international a attribué un chiffre romain à chacune de ces protéines pour éviter toute confusion. Les facteurs associés à l'hémophilie A et B ont respectivement été appelés facteur VIII et facteur IX. Un modèle pour l'interaction de ces facteurs, appelé série de réactions en cascade dans la coagulation sanguine, a été publié en 1964 dans la revue scientifique Nature.
Même si le sang des personnes hémophiles ne coagule pas normalement, ces personnes ne saignent ni plus rapidement ni plus abondamment que d'autres, mais elles saignent plus longtemps.
Même en présence d'hémophilie grave, la coagulation se produit quand même, mais elle est beaucoup plus lente. En général, le caillot ne peut résister à la pression exercée par le sang qui s'échappe. Le caillot est donc constamment perturbé et ne peut se solidifier, ce qui nuit à la cicatrisation de la plaie.
